Abstract: We propose a new therapeutic strategy for Alzheimer's disease (AD). Brain peptide p3-Alcβ37 is generated from the neuronal protein alcadein β through cleavage of γ-secretase, similar to the generation of amyloid β (Aβ) derived from Aβ-protein precursor/APP. Neurotoxicity by Aβ oligomers (Aβo) is the prime cause prior to the loss of brain function in AD. We found that p3-Alcβ37 and its shorter peptide p3-Alcβ9-19 enhanced the mitochondrial activity of neurons and protected neurons against Aβo-induced toxicity. This is due to the suppression of the Aβo-mediated excessive Ca2+ influx into neurons by p3-Alcβ. Successful transfer of p3-Alcβ9-19 into the brain following peripheral administration improved the mitochondrial viability in the brain of AD mice model, in which the mitochondrial activity is attenuated by increasing the neurotoxic human Aβ42 burden, as revealed through brain PET imaging to monitor mitochondrial function. Because mitochondrial dysfunction is common in the brain of AD patients alongside increased Aβ and reduced p3-Alcβ37 levels, the administration of p3-Alcβ9-19 may be a promising treatment for restoring, protecting, and promoting brain functions in patients with AD.
Hata S, Saito H, Kakiuchi T, et al. EMBO Mol Med. Published online March 30, 2023:e17052
Abstract: Alzheimer’s disease (AD) is a very common neurodegenerative disorder, chiefly caused by increased production of neurotoxic β-amyloid (Aβ) peptide generated from proteolytic cleavage of β-amyloid protein precursor (APP). Except for familial AD arising from mutations in the APP and presenilin (PSEN) genes, the molecular mechanisms regulating the amyloidogenic processing of APP are largely unclear. Alcadein α/calsyntenin1 (ALCα/CLSTN1) is a neuronal type I transmembrane protein that forms a complex with APP, mediated by the neuronal adaptor protein X11-like (X11L or MINT2). Formation of the ALCα–X11L–APP tripartite complex suppresses Aβ generation in vitro, and X11L-deficient mice exhibit enhanced amyloidogenic processing of endogenous APP. However, the role of ALCα in APP metabolism in vivo remains unclear. Here, by generating ALCα-deficient mice and using immunohistochemistry, immunoblotting, and co-immunoprecipitation analyses, we verified the role of ALCα in the suppression of amyloidogenic processing of endogenous APP in vivo. We observed that ALCα deficiency attenuates the association of X11L with APP, significantly enhances amyloidogenic β-site cleavage of APP, especially in endosomes, and increases the generation of endogenous Aβ in the brain. Furthermore, we noted amyloid plaque formation in the brains of human APP-transgenic mice in an ALCα-deficient background. These results unveil a potential role of ALCα in protecting cerebral neurons from Aβ-dependent pathogenicity in AD.
Gotoh N, Saito Y, Hata S, et al. Amyloidogenic processing of amyloid β protein precursor (App) is enhanced in the brains of alcadein α–deficient mice. Journal of Biological Chemistry. 2020;295(28):9650-9662.
Introduction: Neuronal p3-Alcβ peptides are generated from the precursor protein Alcadein β (Alcβ) through cleavage by α- and γ-secretases of the amyloid β (Aβ) protein precursor (APP). To reveal whether p3-Alcβ is involved in Alzheimer's disease (AD) contributes for the development of novel therapy and/or drug targets. Methods: We developed new sandwich enzyme-linked immunosorbent assay (sELISA) systems to quantitate levels of p3-Alcβ in the cerebrospinal fluid (CSF). Results: In monkeys, CSF p3-Alcβ decreases with age, and the aging is also accompanied by decreased brain expression of Alcβ. In humans, CSF p3-Alcβ levels decrease to a greater extent in those with AD than in age-matched controls. Subjects carrying presenilin gene mutations show a significantly lower CSF p3-Alcβ level. A cell study with an inverse modulator of γ-secretase remarkably reduces the generation of p3-Alcβ37 while increasing the production of Aβ42. Discussion: Aging decreases the generation of p3-Alcβ, and further significant decrease of p3-Alcβ caused by aberrant γ-secretase activity may accelerate pathogenesis in AD.
Hata S, Omori C, Kimura A, et al. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2019;5(1):740-750.
| Catalog# | Product | Standard Size | Price |
|---|---|---|---|
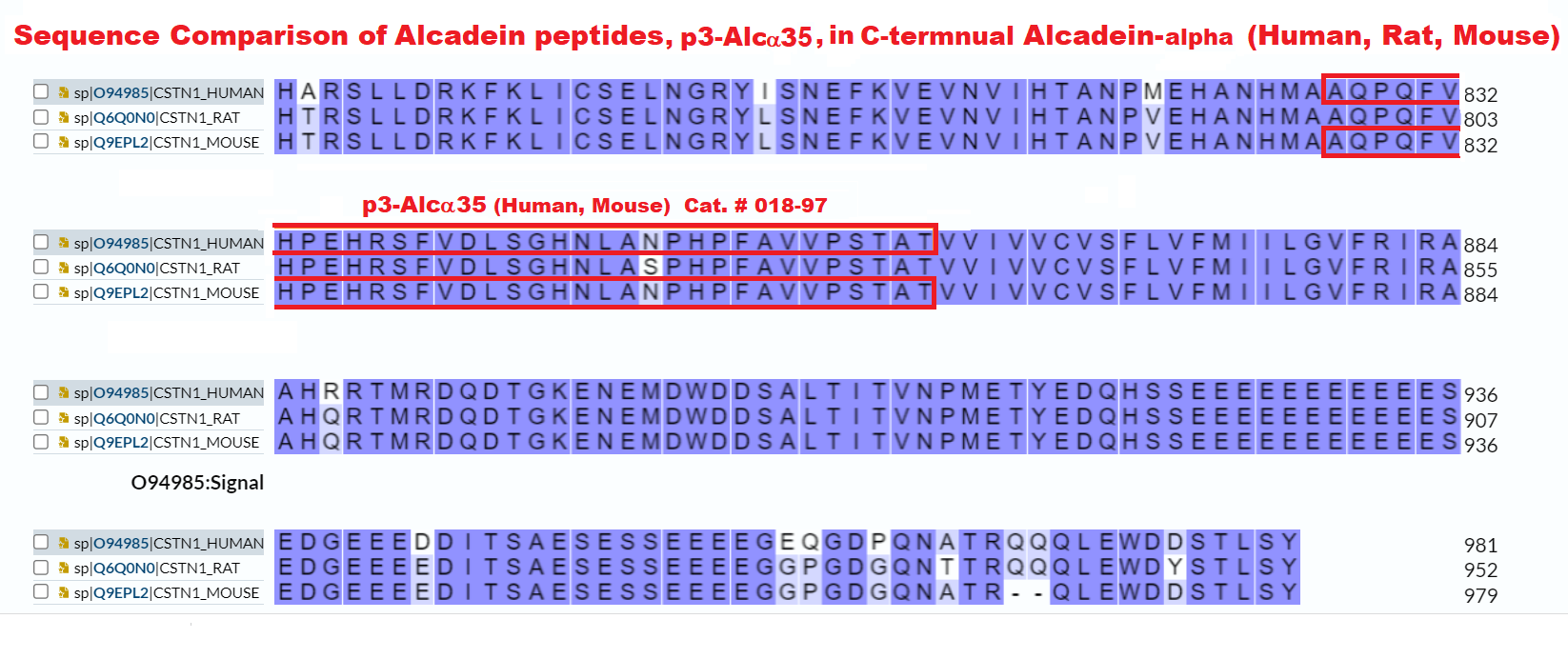
| 018-97 | P3-Alcα35(Human, Mouse) | 100 μg | $310 |
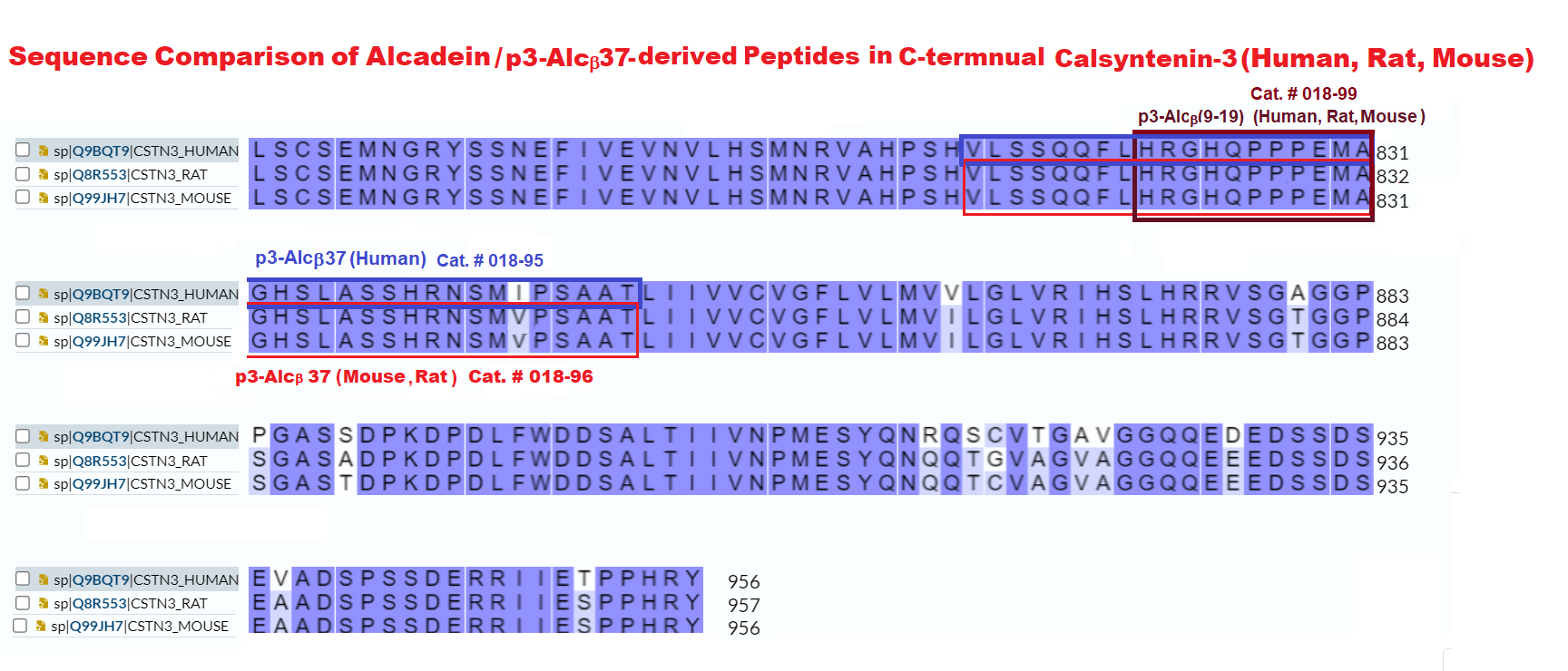
| 018-99 | P3-Alcβ (9-19) (Human, Rat, Mouse) | 200 μg | $116 |
| 018-95 | P3-Alcβ37 (Human) | 100 μg | $320 |
| 018-96 | P3-Alcβ37 (Rat, Mouse) | 100 μg | $320 |
Social Network Confirmation